The molocule DNA contains the genetic instructions used in the development and functioning of all known living organisms. DNA is often compared to a set of blueprints or a recipe, or a code, since it contains the instructions needed to construct other components of cells.
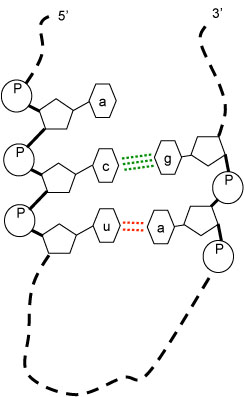
The information in DNA is held in the sequence of the repeating units along the DNA chain, called nucleotides (or "bases"). Certain subsequences of the DNA are genes. When a gene is active (or "expressed"), the information in the gene is copied into a molocule called RNA.
RNA is very much like DNA, but with two important differences:
RNA likes to stick to itself because each one of the bases is attracted to exactly one other bases. The pairs of mutually attractive bases are A-U and G-C. In essence, the RNA twists and folds in order to try to maximize the number of matching pairs that are right across from each other. The particular shape of the folded RNA helps determine its function. For example, one particular shape of RNA could trigger other chemical reactions that make the cell elongate, while a different shape of RNA might trigger reactions that make the cell contract.
The problem of RNA Secondary Structure Prediction is: given an RNA sequence, predict how it will fold. One task within this problem is to determine the number of matching pairs between two aligned subsequences of RNA. For example, suppose the RNA molocule is A-C-G-U-U-U-A-C-U-U, and we want to determine the number of matching pairs between the subsequence A-C-G-U (the beginning of the molocule) and the subsequence U-U-C-A (the tail of the molocule, aligned in reverse). We would check:
We conclude that the number of matching pairs in the two sequences is 3.
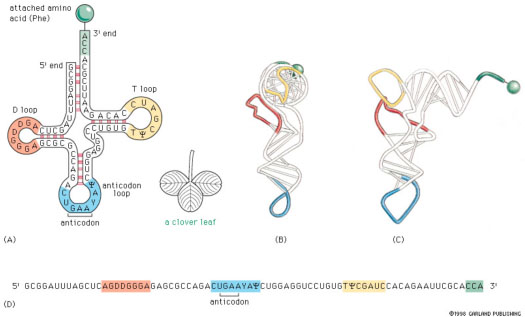
In addition to counting the matches between aligned subsequences of RNA, solving the full secondary structure prediction problem requires guessing which subsequences to try to align, and choosing a set of alignments that yield the most matches. (In general, RNA will stick to itself in several places.) We will not try to solve this (much harder) problem in this class. Below is a small example of a structure containing 4 pairs of alignments. The expression "secondary struture" is used because it is a 2-dimensional model of the RNA. Once secondary structure is predicted, further computations can be used to predict the full 3-dimensional structure of the folded RNA, as shown in (B) and (C) in the figure below.
Working in small groups, develop algorithms for the following two functions. Start with a high-level description of how the algorithm will work (i.e. in pseudocode), and refine it towards something like Python. Don't worry about very minor syntactic details - you won't be actually entering the code into a computer during the workshop. However, you do want to get the logic of the algorithms correct.
MatchSequences( Sequence1, Sequence2 )
This function takes as input two RNA subsequences. Each sequence is represented by a list of numbers, where 0=A, 1=G, 2=U, 3=C. It prints out the number of matching base pairs. For example, calling
MatchSequences( [0,1,0,3], [2,3,2,0] )
should return 3. Recall from your reading of Sec. 11.1-2 of the textbook that if a list is stored in a variable, that you can address individual elements of the list by using square brackets []. For example, if
s = [1,0,3,2]
then s[0]==1, s[1]==0, s[2]==3, and s[3]==2. The length of a list can be determined by using the expression len(<list>). For example, with s specified as above, len(s) == 4.
MatchRNA( RNA, Start1, End1, Start2, End2 )
This function takes as input an RNA molecule specified as a list of number, and counts the matching pairs in two subsequences of the RNA. The first subsequence goes from RNA[Start1] through (and including) RNA[End1], and the second subsequences goes form RNA[Start2] through RNA[End2]. You can assume that the two subsequences will contain the same number of elements. For example,
MatchRNA( [1,0,0,2,2,2,3], 0, 2, 4, 6 )
will print 1, because RNA[0] does not pair with RNA[4], RNA[1] pairs with RNA[5], and RNA[2] does not pair with RNA[6].
MatchRNA has one further complication. If the value of Start2 is greater than End2, then the second subsequence is matched in reverse. For example,
MatchRNA( [1,0,0,2,2,2,3], 0, 2, 6, 4 )
will print 3, because RNA[0] pairs with RNA[6], RNA[1] pairs with RNA[5], and RNA[2] pairs with RNA[4].
After each group develops their pseudocode, they should take turns presenting their solution to the other groups. Everyone attending the lab should be sure to take home their own notes detailing the algorithm their group created. (It is okay for a group to modify its algorithm after seeing what the other groups came up with.)
Working with your partner from the previous assignment, implement, debug, and test an algorithm for MatchRNA. If you and your partner were in different workshops or workshop groups, choose either one of the algorithms you developed in workshop.
Test your implementation on the following data, and use it to fill out the following table:
RNA1 = [0,2,1,2,1,1,1,2,3,3,0,3,1,0,3,2,3,1,2,3,1,2,3,2,0,3,2,0,1,0]
RNA2 = [3,2,1,0,3,1,0,3,2,0,2,0,0,1,1,1,2,3,0,0,1,3]
RNA Start1 End1 Start2 End2 Number Matches ------------------------------------------------------------------- RNA1 3 14 28 17 | ------------------------------------------------------------------- RNA1 3 14 17 28 | ------------------------------------------------------------------- RNA2 4 10 15 21 | ------------------------------------------------------------------- RNA2 4 10 21 15 | -------------------------------------------------------------------
Save your code in a file named RNA.py. Save your table of results in a file named RNA_results.txt. (You can copy the table above in a new file window in IDLE, edit it to add your results, and save it with the name RNA_results.txt.) Turn in both files using Blackboard no later than 10:00am Saturday September 26. Only turn in one copy for a pair of partners. Include the names of both partners in the "Comments" box when you upload the files with Blackboard.
The way RNA folds can be more precisely characterized as a process by which the molecule tries to reach a state of "minimum energy". Every time matching base pairs align, the energy of the molecule goes down. However, whenever a series of matching pairs stops matching up, there is a "bulge" which causes the energy to go up. The problem of RNA Secondary Structure Prediction can then be stated as, given an RNA sequence, determine a folding that has minimum energy.
A simplified energy model for RNA is as follows. Each matching pair gives -1.8 units of energy. Each mismatching pair gives +3.5 units of energy, however, an adjacent sequence of mismatching pairs is only penalized once, for a total of +3.5. For example, matching [0,1,2,2] with [2,1,1,0] is scored as -1.8 + 3.5 -1.8 for a total of -0.1
Implement a function EnergyRNA(RNA, Start1, End1, Start2, End2) that does this calculation. Test it on the data above. Add a column title "Energy" to the table above, and record the values you calculated. Be sure to indicate in the Comments box when you turn in the assignment that you did the extra credit.